
甲状腺癌(thyroid carcinoma,TC)是内分泌系统 最常见的恶性肿瘤。根据中国肿瘤登记中心的数据显示,中国城市地区女性甲状腺癌发病率位居女性所有恶性肿瘤的第4位。

中国家族遗传性肿瘤临床诊疗专家共识(2021 年版)
(5)-家族遗传性甲状腺癌
致病基因变异
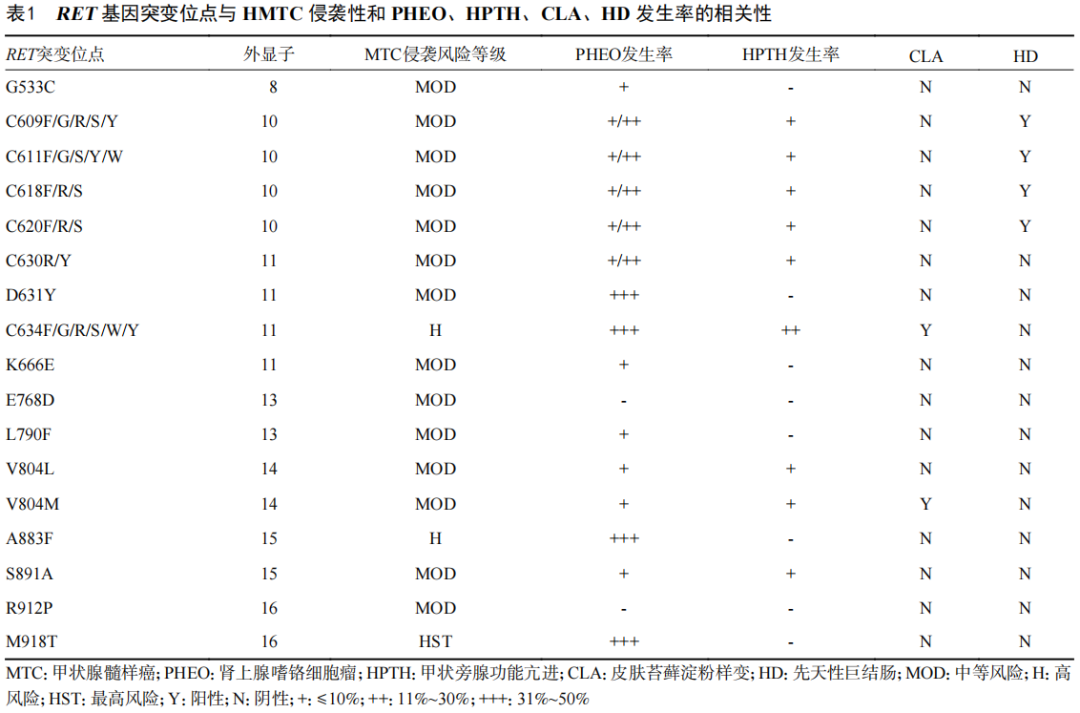
遗传性甲状腺髓样癌(HMTC):约95%的HMTC由RET基因突变导致。95%的MEN2A患者的RET基因在10号外显子的第609、611、618、620位以及11号外显子的第634位密码子发生突变。95%的MEN2B患者携带RET基因第16号外显子M918T突变,不足5%的MEN2B患者携带15号外显子A883F突变。根据ATA指南,HMTC患者的侵袭性风险依RET基因突变位点分为最高风险(HST)、高风险(H)和中等风险(MOD)。
家族性甲状腺非髓样癌(FNMTC):目前尚未发现FNMTC的特异性致病基因。综合征型FNMTC与已知的肿瘤易感基因相关,如APC基因是FAP综合征的致病基因。而在非综合征型FNMTC致病基因的研究中,已发现DICER1、FOXE1、PTCSC2、POT1、TCO、NMTC1等基因与FNMTC的易感性相关,亦有发现FNMTC与14q、19p、2q、1q、8p、6q和12q突变有关,但均缺乏可重复性。FNMTC没有热点基因变异,涉及的基因也比较多。因此,对于FNMTC可以考虑广泛的多基因筛查。
检测建议
基因检测包括胚系和体细胞变异检测,对于家族遗·传性甲状腺癌主要进行胚系突变的检测,临床推荐对外周血进行检测。HMTC患者或高危人群主要检测RET基因胚系变异,临床常用的检测方法包括聚合酶链式反应(polymeRASe chain reaction,PCR)、一代测序技术(Sanger sequencing)和二代测序技术(next generation sequencing,NGS)。不同的FNMTC家系基因变异位点可能不同,推荐 FNMTC 患者或高危人群采用NGS进行较为广泛的基因胚系突变筛查,先证者直系家属可采用一代测序技术进行验证。
前言
新近研究表明中国每年新发肿瘤患者约457万例,其中家族遗传性肿瘤约 5%~10%。家族遗传性肿瘤具有发病早、多原发病灶、家族聚集等特点,其发病机制、临床表型、诊疗策略及家系管理不同于散发性肿瘤。目前国内尚缺乏关于家族遗传性肿瘤临床诊疗的共识及指南。基于此,中国抗癌协会家族遗传性肿瘤专业委员会组织国内临床一线专家共同制定了《中国家族遗传性肿瘤临床诊疗专家共识》,主要涵盖家族遗传性乳腺癌、卵巢癌、胃癌、结直肠癌、甲状腺癌、肾癌和前列腺癌七个部分,连载刊出。专业委员会前期也推出了“中国家族遗传性肿瘤基因突变数据库(CFCSG-database,http://cfcsg-database.org.cn/)”。该数据库主要储存和分析中国家族遗传性肿瘤相关易感基因胚系突变数据,尤其是中国人群特有或新发现的突变。本共识及相关数据库将为中国家族遗传性肿瘤遗传咨询、风险评估、预防干预及临床治疗提供参考。
共识内容
甲状腺癌(thyroid carcinoma,TC)是内分泌系统最常见的恶性肿瘤。根据中国肿瘤登记中心的数据显示,中国城市地区女性甲状腺癌发病率位居女性所有恶性肿瘤的第4位。家族遗传性甲状腺癌包括遗传性甲状腺髓样癌(hereditary medullary thyroidcarcinoma,HMTC)和家族性甲状腺非髓样癌(familial non-medullary thyroid carcinoma,FNMTC)。
1 遗传性甲状腺髓样癌
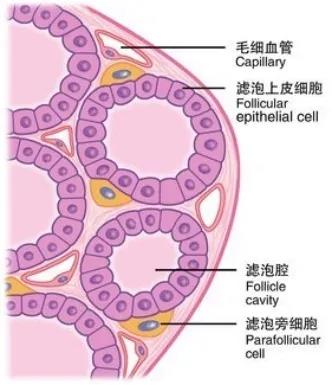
甲状腺髓样癌(medullary thyroid carcinoma,MTC)是来源于甲状腺滤泡旁细胞(C细胞)的恶性肿瘤,约占甲状腺恶性肿瘤的5%,甲状腺滤泡旁细胞具有合成分泌降钙素(calcitonin,Ctn)及降钙素基因相关肽的作用。因此,MTC亦被认为是神经内分泌肿瘤之一。在MTC中25%~30%为HMTC。根据ATA指南(TheAmerican Thyroid Association),HMTC可以分为多发性内分泌肿瘤2A型(MEN2A)和多发性内分泌肿瘤2B型(MEN2B)。
MEN2A型HMTC根据临床表现的差异可分为4种类型:经典型MEN2A、MEN2A伴皮肤苔藓淀粉样变(cutaneous lichen amyloidosis,CLA)、MEN2A伴先天性巨结肠(Hirschsprung's disease,HD)、家族性MTC(FMTC)。MEN2B型以MTC并发黏膜多发性神经瘤为特点,通常在婴儿期发病,且具有较高的侵袭性,早期即可发生淋巴结甚至远处转移。
2 家族性甲状腺非髓样癌
甲状腺非髓样癌(non-medullary thyroid carci-noma,NMTC)包括甲状腺乳头状癌(papillary thyroid carcinoma,PTC)、甲状腺滤泡癌(follicular thyroid carcinoma,FTC)和甲状腺未分化癌(anaplastic thyroid carcinoma,ATC)等起源于甲状腺滤泡上皮细胞的恶性肿瘤,约占所有TC的90%,其中PTC是最常见的病理类型。5%~10%的NMTC患者表现为家族聚集性。
FNMTC的定义是家族一级亲属间具有2个或2个以上的NMTC患者并排除头颈部射线暴露史。FNMTC可分为2类:
1)以非甲状腺肿瘤为主要表现的家族性肿瘤综合征,包括家族性腺瘤性息肉病(familial adenomatous polyposis,FAP)、Cowden综合征等[1],而FNMTC在此类遗传综合征中可能是首发表现[2];
2)非综合征相关,在一个患病家系中患者以发生NMTC为特征,不合并其他内分泌肿瘤或疾病。FNMTC生物学行为呈现高侵袭性,如患者发病年龄早,多灶、双侧发病比例高,局部浸润,淋巴结转移率高,复发率高,无病生存期短[3]等。
3风险评估及基因检测
3.1风险评估及适检人群
3.1.1风险评估
1)HMTC:约95%的HMTC由RET基因突变导致。95%的MEN2A患者的RET基因在10号外显子的第609、611、618、620位以及11号外显子的第634位密码子发生突变[4]。95%的MEN2B患者携带RET基因第16号外显子M918T突变,不足5%的MEN2B患者携带15号外显子A883F突变[5]。根据ATA指南,HMTC患者的侵袭性风险依RET基因突变位点分为最高风险(HST)、高风险(H)和中等风险(MOD)[6],见表1。
2)FNMTC:尚未发现FNMTC的特异性致病基因,因此无法根据某个特定基因的变异特征对FNMTC患者进行危险分层。在中国FNMTC家系研究中,发现不同FNMTC家系中具有不同的肿瘤易感基因变异,包括APC、MSH6、BRCA1/2基因等[7]。不同基因突变导致的TC发病风险不同,如携带PTEN胚系突变的患者中约1/4伴发TC,且发病年龄早,FTC比率高[1]。上述易感基因变异与其他肿瘤的发生亦有相关性,提示罹患第二种肿瘤的风险增加。如携带MSH6致病突变的FNMTC患者发生结直肠癌和子宫内膜癌的风险增高。
3.1.2适检人群
临床上1%~7%的散发性MTC患者实际具有HMTC的遗传背景,因此散发性病例行基因筛查可进一步明确疾病分型。MTC基因筛查有助于在家系成员中排查特定种类的HMTC,并根据不同的突变位点进行危险分层,有针对性地制定治疗策略。
鉴于NMTC(尤其PTC)的高发病率,很多散发性甲状腺非髓样癌(sporadicnon-medullarythyroidcarcinoma,SNMTC)也可能存在家系聚集现象。相比SNMTC,FNMTC患者罹患第二种肿瘤风险增加,且可能成为致死原因。因此对有家族史的NMTC患者进行遗传易感基因的筛查有助于FNMTC和SNMTC的鉴别,也有助于对NMTC临床预后和继发肿瘤风险的评估。
专家组意见:推荐对以下人群进行RET基因筛查:有家族史的MTC患者本人及一级亲属;在儿童或婴儿期出现MEN2B表现的患者本人及其父母;皮肤苔藓淀粉样变患者;先天性巨结肠病患者;肾上腺嗜铬细胞瘤的患者;患有SNMTC且有检测意愿者。推荐对有家族史的NMTC患者及其一级亲属进行肿瘤易感基因筛查。

3.2检测基因
1)HMTC:RET基因突变是MTC发病的主要分子病因学基础。
2)FNMTC:目前尚未发现FNMTC的特异性致病基因。综合征型FNMTC与已知的肿瘤易感基因相关,如APC基因是FAP综合征的致病基因[8]。而在非综合征型FNMTC致病基因的研究中,已发现DICER1、FOXE1、PTCSC2、POT1、TCO、NMTC1等基因与FNMTC的易感性相关[9-11],亦有发现FNMTC与14q、19p、2q、1q、8p、6q和12q突变有关[12-13],但均缺乏可重复性。FNMTC没有热点基因变异,涉及的基因也比较多。因此,对于FNMTC可以考虑广泛的多基因筛查。
3.3检测方法
基因检测包括胚系和体细胞变异检测,对于家族遗传性甲状腺癌主要进行胚系突变的检测,临床推荐对外周血进行检测。HMTC患者或高危人群主要检测RET基因胚系变异,临床常用的检测方法包括聚合酶链式反应(polymeRASe chain reaction,PCR)、一代测序技术(Sanger sequencing)和二代测序技术(next generation sequencing,NGS)。不同的FNMTC家系基因变异位点可能不同,推荐FNMTC患者或高危人群采用NGS进行较为广泛的基因胚系突变筛查,先证者直系家属可采用一代测序技术进行验证。
3.4检测结果解读
HMTC相关的RET基因胚系突变以及FNMTC相关的基因胚系突变可参考国际癌症研究机构(IARC)分类,根据变异的致病性分为5类:5类:致病性;4类:可能致病性;3类:意义未明;2类:可能良性;1类:良性。数据解读的标准和规范以及数据解读和注释流程可参考《美国医学遗传学与基因组学学会(ACMG)和美国分子病理学会(AMP)序列变异解读标准和指南(2015版)》[14-15]。临床生物信息分析团队主要由具有 临床医学、分子生物学、生物信息学、遗传学和检验学等专业背景的人员组成,需经生物信息学培训和适当的资质确认。标准肿瘤基因变异检测报告应包含检测方法、检测范围、检测结果以及结果解读。
专家组意见:可疑HMTC患者或高危人群胚系RET基因检测,可考虑使用PCR、一代测序和NGS方法;可疑FNMTC患者或高危人群可采用NGS进行广泛的多基因筛查;已知突变者的直系家属可采用一代测序技术进行验证。应由具有专业背景并经系统培训和资质确认的临床生物信息分析员进行数据质控、基因变异分类和解读。
4诊断策略
4.1HMTC的检测
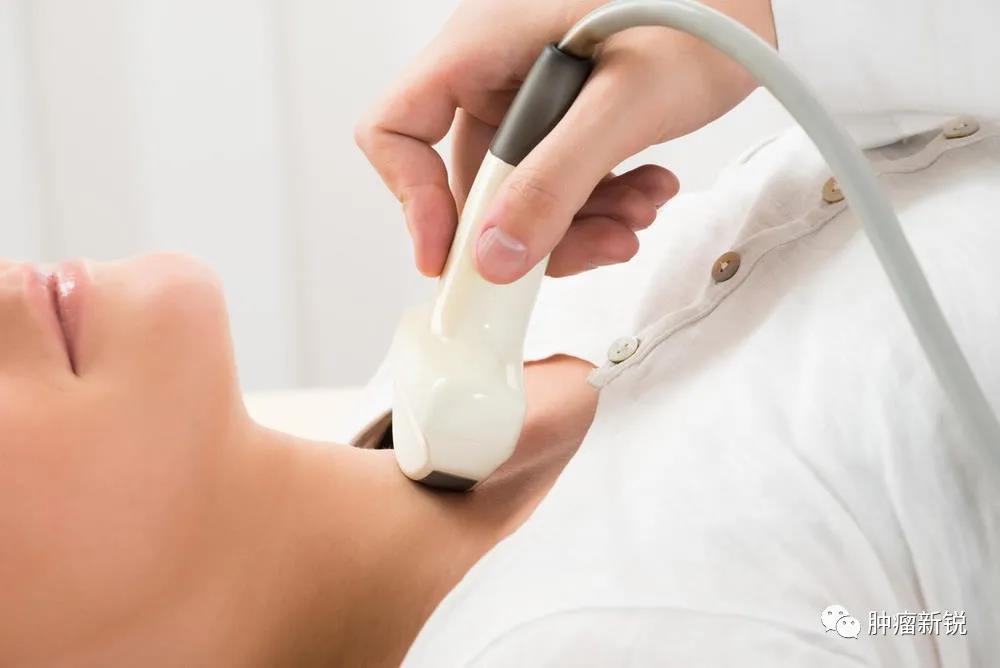
4.1.1超声检查及超声引导下细针抽吸活检
超声检查是MTC检测的首选影像学方法,可实时评估甲状腺病灶的良恶性及其与周围组织的关系,同时对颈部淋巴结进行评估。TI-RADS分类基于超声特征对甲状腺结节进行评分及分类,评估恶性风险,指导结节管理。绝大多数MTCTI-RADS分类为4类或5类,表明超声检查对MTC具有较高的诊断价值[16-17]。由于MTC细胞学形态表现多样且缺乏淀粉样蛋白,超声检查及超声引导下细针抽吸活检(fine needle aspiration,FNA)诊断MTC准确率约50%[18]。FNA标本中肿瘤细胞数足够时,免疫组织化学染色可以提高诊断准确性,诊断标准包括降钙素、癌胚抗原、嗜铬粒蛋白染色阳性及甲状腺球蛋白缺失。
专家组意见:超声检查和TI-RADS对MTC具有较高的诊断价值,约50%的MTC可通过FNA获取细胞病理学诊断。推荐超声检查联合FNA用于HMTC筛查,超声TI-RADS分类评估恶性风险,必要时推荐免疫组织化学染色。
4.1.2血清降钙素检测
血清降钙素是MTC中灵敏度及特异度均较高的肿瘤标志物。血清降钙素检测比其他MTC诊断方法的临床分期早,预后好,因此欧洲学者建议甲状腺结节常规检测血清降钙素用于MTC早期诊断[19]。但MTC的低发病率引发了对常规检测成本效益的关注[20],且非分泌型MTC不分泌降钙素,故美国学者对血清降钙素常规检测持中立态度。目前血清降钙素诊断MTC的阈值尚未统一。基于大样本研究,一般认为血清降钙素<10pg/mL时基本排除MTC;=100pg/mL时高度怀疑MTC;10~100pg/mL时可疑MTC,需联合降钙素激发试验和细针抽吸洗脱液降钙素检测排除非MTC血清降钙素升高[21]。
专家组意见:血清降钙素是MTC特异性和敏感度较高的肿瘤标志物,其升高提示MTC可能,但甲状腺结节常规检测血清降钙素还存在争议。临床可疑HMTC推荐进行血清降钙素检测,当血清降钙素10~100pg/mL时,推荐联合降钙素激发试验和细针抽吸洗脱液降钙素检测。
4.1.3基因检测及其他检测
HMTC的遗传基础为RET基因胚系变异。详细内容见前文“风险评估及基因检测”。HMTC局部侵犯范围广、颈部淋巴结转移和远处转移率高。除颈部超声及血清降钙素检测,术前评估还需结合其他检查。血清CEA水平反映肿瘤侵犯范围及分化程度,可用于评估肿瘤侵袭性。CT评估中央区、上纵隔和咽后间隙转移性淋巴结以及与周围组织关系要优于超声,CT对肺转移灵敏度最高;强化CT和MRI是检测肝转移的灵敏方法;MRI和骨显像是较为敏感的骨转移检查方法;PET/CT可用于全身转移性病灶的评估,但目前尚无足够的证据支持PET/CT用于HMTC临床分期[22]。
专家组意见:推荐在确诊MTC的基础上,结合RET基因胚系突变检测及家族史来明确HMTC的诊断。对HMTC患者联合颈部超声检查、血清降钙素和CEA检测以综合评估肿瘤侵犯程度及临床进展。临床可疑颈部广泛侵犯及远处转移时,推荐结合颈胸部CT、肝脏强化CT或MRI、骨MRI或骨显像和PET-CT等检查。
4.2FNMTC
4.2.1超声检查
鉴于FNMTC和SNMTC在超声影像特征上差异甚微,目前超声检查在FNMTC中的诊断价值尚存在争议。PTEN胚系突变综合征型FNMTC甲状腺癌的发生风险较大,发病早,FTC比率高,因此建议PTEN胚系突变患者应在诊断时进行甲状腺超声检查。
4.2.2基因检测
FNMTC的诊断主要依靠家族史,FNMTC需符合家族一级亲属间具有2个或2个以上的NMTC患者并排除头颈部射线暴露史。尚未发现FNMTC的特异性致病基因,详细内容见前文“风险评估及基因检测”。
专家组意见:综合征型FNMTC具有较明确的致病基因,具有典型综合征表型的甲状腺癌患者应进行靶向基因检测以确诊FNMTC。而非综合征型FNMTC无特异性致病基因,诊断主要依靠家族史。
5 治疗策略
5.1 针对HMTC的治疗
5.1.1 针对可手术HMTC的治疗
HMTC患者首选手术治疗,传统的放化疗疗效不佳。目前国内外对HMTC原发灶治疗的意见统一,全甲状腺切除术及中央区淋巴结清扫是最基本的手术方式[9,23]。对于中央区淋巴结转移负荷较大者建议行预防性上纵隔清扫;对于上纵隔淋巴结转移的患者,应行治疗性上纵隔淋巴结清扫。对于cN1b患者,应行治疗性侧颈淋巴结清扫;而对于cN0患者是否行预防性侧颈清扫,仍存在争议。有研究表明,侧颈淋巴结转移率与中央区淋巴结转移数量密切相关,1~3枚中央区淋巴结转移时,同侧侧颈淋巴结转移率为77%,当中央区淋巴结转移=4枚时,同侧侧颈转移率可达到98%[24]。术前基础血清Ctn水平也可一定程度上反映淋巴结转移程度[23]。
专家组意见:手术是HMTC的首选治疗方式。对于cN0HMTC患者,推荐在全甲状腺切除的基础上行双侧预防性中央区清扫术。对于cN1aHMTC患者,均应行治疗性中央区淋巴结清扫。对于cN1b HMTC患者,应行治疗性中央区和侧颈淋巴结清扫。结合原发灶、中央区淋巴结转移情况及血清Ctn水平决定是否行预防性侧颈淋巴结清扫。
5.1.2局部晚期不可手术及远处转移性HMTC的治疗
HMTC恶性程度高,部分HMTC患者在初次就诊时即为局部晚期和(或)合并远处转移,R0切除的机会极低,且需要付出多种器官功能丧失的代价。对于上述患者而言,总体治疗目标是提高局部控制率、缓解全身症状与转移灶症状、减少疾病相关死亡。目前已报道多种靶向药物对晚期HMTC有效[25-27]。小分子多靶点受体酪氨酸激酶抑制剂凡德他尼和cabozantinib/ target=_blank class=infotextkey>卡博替尼已被欧美批准用于局部晚期或远处转移性MTC的临床治疗,还有多种多靶点小分子酪氨酸激酶抑制剂正在进行临床试验。除上述多靶点酪氨酸激酶抑制剂之外,还有高选择性RET抑制剂,其对RET的亲和力高,对于RET的融合突变及点突变均有效。目前已有两种小分子高选择性RET抑制剂(BLU-667和LOXO-292)在临床试验阶段[28-29]。
专家组意见:对局部晚期或存在远处转移不适宜手术的HMTC患者可考虑采用靶向药物治疗。医生需要衡量肿瘤生长速度、生存质量与治疗毒性之间的关系,合理选择治疗方案。
5.1.3针对HMTC并发症的治疗
MEN2A型患者通常在30~40岁时出现肾上腺嗜铬细胞瘤(pheochro- mocytoma,PHEO),通常与MTC同时或随后诊断。在ATA-H和ATA-HST类别的患者中,PHEO可能在8~12岁时已出现,在ATA-MOD类别患者中,PHEO可能在19岁时出现[30]。甲状腺切除术时未诊断出PHEO可能导致患者出现严重并发症甚至死亡。因此,对于HMTC患者,术前应仔细排查PHEO,一旦确诊应该先切除PHEO。HMTC确诊后应通过MI-BI显像、超声及CT筛查定位甲状旁腺功能亢进(hyp- erparathyroidism,HPTH),若4个腺体均增生,手术可选择次全甲状旁腺切除术或全甲状旁腺切除术,自体异位移植。
专家组意见:建议HMTC患者尽早筛查PHEO,一旦确诊应该先切除PHEO。HMTC确诊后应通过MIBI显像、超声及CT筛查定位HPTH。HPTH患者应切除明显增大的甲状旁腺,术后给予替代治疗,并监测甲状腺功能。
5.2 针对FNMTC的治疗
FNMTC具有常染色体显性遗传模式,FNMTC一级亲属的发病率较普通人群提高5~8倍,而FN-MTC病例具有高度的遗传分子异质性,难以识别关键的遗传分子变化[31]。由于FNMTC与SNMTC患者的临床特征差异甚微,因此治疗策略基本相同[32]。但与SNMTC相比,FNMTC发病年龄更早、侵袭性更强、多灶性和淋巴结转移率更高、复发更频繁[31,33],因此FNMTC在行手术治疗时需要常规清扫中央区淋巴结,当中央区淋巴结转移较多时,提示可能需要加行侧颈部淋巴结清扫[3,34]。FNMTC倾向于多灶性及双侧发病,所以在为FNMTC患者选择手术术式时,可
能需要适当放宽双侧甲状腺全切除的指征[3,35]。侵犯被膜及周围软组织被认为是评价肿瘤侵袭性的重要指标之一,FNMTC比SNMTC更具有侵袭性,对FNM-TC可能需要增加手术切除范围[34,36]。
专家组意见:目前FNMTC治疗策略和SNMTC患者基本相同,FNMTC更容易发生淋巴结转移,在行手术治疗时需要常规清扫中央区淋巴结,当中央区淋巴结转移较多时,可加行侧颈部淋巴结清扫。
6 高危个体干预
6.1 HMTC的干预
高危个体早期干预应根据不同的突变位点及不同的风险分层进行相应的管理,不能一概而论。对于HST级别患者,应尽早行甲状腺切除术。高度怀疑为HST级别的婴儿出生应立即进行基因检测,若确诊为HST级别,推荐干预时间为出生的第1年内进行甲状腺切除术。对于H级别患者,应从3岁起每年进行体检、颈部超声和血清Ctn检测,在5岁之前进行甲状腺切除术,并根据Ctn水平指导手术时间和手术范围。对于MOD级别患者,应从5岁起每年进行体检、颈部超声和血清Ctn检测,在儿童期或成年期进行甲状腺切除术,手术时间主要取决于血清Ctn水平[9]。因为是预防性手术,且文献资料极少,亦缺乏相关的法律条文指引,建议在与患儿监护人充分沟通后方可参考。
专家组意见:HMTC与RET基因变异存在明显相关性,推荐HMTC患者及其家属尽早进行RET基因检测,有助于评估甲状腺癌遗传风险,对疾病进行准确的危险分层。推荐通过多学科会诊制订合理的早期干预、治疗与随访方案,并与患者或患儿监护人充分沟通,共同决定干预手段和时机。开展预防性甲状腺切除术,应获得所在医院伦理委员会审批通过,必要时需要咨询相关法律。对于有生育需求的 RET基因胚系突变携带者,应告知RET基因胚系突变可能给家庭成员带来的风险,建议进行孕前或产前遗传咨询。
6.2 FNMTC的干预
FNMTC家系还存在“遗传早现”的现象,遗传早现是指某种遗传病在连续世代中,发现其症状一代比一代严重,而发病时间一代早于一代[3]。因此,推荐对无症状或无可触及结节的FNMTC家族成员定期进行甲状腺功能血清学检测及颈部超声筛查,以期及时发现,争取较好的治疗效果。对所有NMTC患者均需详细询问家族史,若发现家族成员中有2例或2例以上NMTC患者,应对其所有20岁以上的一级和二级亲属,尤其是女性,进行1次/年的甲状腺B超扫描筛查。对腺瘤样甲状腺肿合并多发NMTC患者,即使无甲状腺癌家族史,也建议行家族性筛查。
专家组意见:对无症状或无可触及结节的FNMTC家族成员,要定期进行甲状腺功能血清学检测及颈部超声筛查,以期及时发现,早期治疗,争取较好的治疗效果。此外推荐FNMTC患者及其家属等高危个体进行全面甲状腺癌遗传易感相关的多基因检测,有助于评估甲状腺癌遗传风险,制订合理的治疗与随访方案。
参考文献
[1]Peiling Yang S, Ngeow J. Familial non-medullary thyroid cancer: unraveling the genetic maze[J]. Endocr Relat Cancer, 2016, 23(12): R577-R595.
[2]Stulp RP,Herkert JC,Karrenbeld A, etal. Thyroid cancer in apatient with a germline MSH2 mutation. Case report and review of the Lynch syndrome expanding tumour spectrum[J]. HeredCan- cer Clin Pract, 2008, 6(1):15.
[3]CapezzoneM,MarchisottaS, CantaraS, etal.Familialnon- medullarythyroid carcinoma displays the features of clinicalanti- cipation suggestive of a distinctbiological entity[J]. Endocr Relat Cancer, 2008, 15(4):1075-1081.
[4]Raue F, FrankRaue K. Genotypephenotype cORRelation inmul- tiple endocrine neoplasia type 2[J]. Clinics (Sao Paulo), 2012, 67(Suppl 1):69-75.
[5]Smith DP, Houghton C, Ponder BA. Germline mutationofRET Codon 883 in two cases of de novo MEN 2B[J]. Oncogene, 1997, 15(10):1213-1217.
[6]WellsSAJr, Asa SL, Dralle H, et al. Revised American thyroid asso- ciationguidelines for the management of medullary thyroid car- cinoma[J]. Thyroid, 2015, 25(6):567-610.
[7]YuY, Dong L, Li DP, et al. Targeted DNA sequencing detects muta- tions related to susceptibility among familial non-medullary thyroid cancer[J]. Sci Rep, 2015, 5:16129.
[8]Cetta F, Montalto G, Gori M, et al. Germline mutations ofthe APC gene in patients with familial adenomatous polyposisassociated thyroid carcinoma: resultsfrom a European cooperative study[J]. J Clin Endocrinol Metab, 2000, 85(1):286-292.
[9]Hińcza K, Kowalik A,Kowalska A.Current knowledgeof germline genetic risk factors for the development of non-medullary thyroid cancer[J]. Genes (Basel), 2019, 10(7):E482.
[10]Srivastava A, Miao BP, Skopelitou D, et al. A germline mutation in the POT1 gene is a candidate for familial non-medullary thyroid cancer[J]. Cancers (Basel), 2020, 12(6):E1441.
[11]McKay JD, ThompsonD,Lesueur F, et al. Evidence for interaction between the TCO and NMTC1 loci in familial non-medullary thyroid cancer[J]. J Med Genet, 2004, 41(6):407-412.
[12]Navas-Carrillo D, Ríos A, Rodríguez JM, et al. Familial nonmed- ullary thyroid cancer: screening, clinical, molecular and genetic findings[J]. Biochim Biophys Acta, 2014, 1846(2):468-476.
[13]Capezzone M, Robenshtok E, CantaraS,et al. Familialnon- medullary thyroid cancer: a critical review[J]. J Endocrinol Invest, 2021, 44(5):943-950.
[14]Plon SE, Eccles DM, Easton D, et al. Sequencevariant classification and reporting: recommendations for improving the interpretation of cancer susceptibility genetic test results[J]. Hum Mutat, 2008, 29(11):1282-1291.
[15]罗定远,廖健伟 . 甲状腺癌基因检测与临床应用广东专家共识 (2020版)[J]. 中华普通外科学文献(电子版),2020,14(3):161-168.
[16]Li JM, Li HR, Yang Y, et al. The KWAK TI-RADS and 2015 ATA gui- delines for medullary thyroid carcinoma: combined with cell block- assisted ultrasound-guided thyroid fine-needle aspiration[J]. Clin Endocrinol (Oxf), 2020, 92(5):450-460.
[17]ZhaoJ, Yang F, Wei X, etal. Ultrasound features value in the diagnosis and prognosis of medullary thyroid carcinoma[J]. Endo- crine, 2021, 72(3):727-734.
[18]TrimboliP,TregliaG,GuidobaldiL, etal. DetectionrateofFNA cytology in medullary thyroid carcinoma: a meta-analysis[J]. Clin Endocrinol (Oxf), 2015, 82(2):280-285.
[19]Niederle B. Screeningfor medullary carcinoma ofthe thyroid[J]. Br JSurg, 2014, 101(13):1625-1626.
[20]Verbeek HH, de Groot JWB, Sluiter WJ, et al. Calcitonin testing for detectionof medullary thyroid cancer in people with thyroid nod- ules[J]. Cochrane Database Syst Rev, 2020, 3(3):CD010159.
[21]Giannetta E, Guarnotta V, Altieri B, et al. ENDOCRINE TUMOURS: Calcitonin in thyroidandextrathyroid neuroendocrine neoplasms: thetwofacedJanus[J].EurJ Endocrinol, 2020, 183(6):R197-R215.
[22]Giovanella L, Treglia G, Iakovou I, et al. EANM practice guideline for PET/CT imaging in medullary thyroid carcinoma[J]. Eur J Nucl Med Mol Imaging, 2020, 47(1):61-77.
[23]王宇, 田文,嵇庆海,等 . 甲状腺髓样癌诊断与治疗中国专家共识 (2020版)[J]. 中国实用外科杂志,2020,40(9):1012-1020.
[24]WeberT, SchillingT, FrankRaue K, et al. Impact of modified radic- al neck dissection on biochemical cure in medullary thyroid car- cinomas[J]. Surgery, 2001, 130(6):1044-1049.
[25]Tappenden P, Carroll C, Hamilton J, et al. cabozantinib andvan- detanib for unresectable locally advanced or metastatic medullary thyroid cancer: a systematic review and economic model[J]. Heal- th TechnolAssess, 2019, 23(8):1-144.
[26]Ruan XH, Shi XL, Dong QM, et al. Antitumor effects of anlotinib inthyroid cancer[J]. Endocr Relat Cancer, 2019, 26(1):153-164.
[27]Chen J, Ji Q, Bai C, et al. Surufatinib in chinese patients with locally advanced or metastatic differentiatedthyroid cancer and med- ullarythyroid cancer: a multicenter, open-label, phase Ⅱ trial[J]. Thyroid. 2020, 30(9): 1245-1253.
[28]Drusbosky LM, Rodriguez E, Dawar R, et al. Therapeutic strategies in RET gene rearranged non-small cell lung cancer[J]. J Hematol Oncol, 2021, 14(1):50.
[29]SubbiahV, Hu MI, Wirth LJ, et al. Pralsetinib for patients with ad- vanced or metastatic RET-altered thyroid cancer (ARROW): a multi- cohort, open-label, registrational, phase 1/2 study[J]. LancetDia-betes Endocrinol, 2021, 9(8):491-501.
[30]Rowland KJ, Chernock RD, Moley JF. Pheochromocytoma in an 8- year-old patient with multiple endocrine neoplasia type 2A: im- plications for screening[J]. J Surg Oncol, 2013, 108(4):203-206.
[31]AmmarSA, Alobuia WM, Kebebew E. An update on familial nonmedullarythyroidcancer[J]. Endocrine, 2020, 68(3):502-507.
[32]Ito Y,Hirokawa M, Higashiyama T, etal.Biological behavior of papillarycarcinomaof the thyroid including squamous cell car- cinoma componentsand prognosis of patients who underwent locally curative surgery[J]. J Thyroid Res, 2012, 2012:230283.
[33]El Lakis M, Giannakou A, Nockel PJ, et al. Do patients with familial nonmedullarythyroid cancer present withmore aggressive dis- ease? Implications for initial surgical treatment[J]. Surgery, 2019, 165(1):50-57.
[34]Mazeh H, BenavidezJ, Poehls JL, et al. In patientswiththyroid can- cer offollicular cell origin, afamily history of nonmedullarythyroid cancer in onefirstdegree relative is associated with more aggress- ive disease[J]. Thyroid, 2012, 22(1):3-8.
[35]Capezzone M, Fralassi N, Secchi C, et al. Longterm clinical out- come in familial and sporadic papillary thyroid carcinoma[J]. Eur Thyroid J, 2020, 9(4):213-220.
[36]ParkYJ, Ahn HY, ChoiHS,etal. The longterm outcomes of the second generation offamilial nonmedullary thyroid carcinoma are more aggressive than sporadic cases[J]. Thyroid, 2012, 22(4):356- 362.